
Opened 9 years ago
Last modified 8 years ago
#887 closed task
Release export wizard — at Version 34
| Reported by: | Nicklas Nordborg | Owned by: | Nicklas Nordborg |
|---|---|---|---|
| Priority: | critical | Milestone: | Reggie v4.5 |
| Component: | net.sf.basedb.reggie | Keywords: | |
| Cc: |
Description (last modified by )
Implement a wizard for creating all files that should be included in a release. Th wizard should take an item list with raw bioassays as input and produce a lot of files. Unless noted, all files are tab-separated text files.
- Transcript data (in folder
dataTables/transcriptDataTable):tidmatrix.features.txt: Array design features with some annotations. The first line is a header line:id,geneSymbol,refSeq,protAcc,description,chr,entrez.- Rows are sorted by external ID (or internal ID?? see comment below for more information)
- All raw bioassays in the input list must use the same array design.
tidmatrix_data.txt: FPKM values for all raw bioassays. Each row represents a feature and each column a raw bioassay.- The first line is a header line with raw bioassay names.
- The first column contains the feature ID.
- Same order of rows as the
tidmatrix.features.txt.
tidmatrix_FPKM_conf_hi.txt,tidmatrix_FPKM_conf_lo.txt,tidmatrix_FPKM_status.txt: More data files similar to thetidmatrix_data.txtfile but with theFPKM_conf_hi,FPKM_conf_loandFPKM_statusvalues.
- Gene data (in folder
dataTables/geneDataTable):genematrix_data.txt: Sum of FPKM values per gene symbol.- The first line is a header line with raw bioassay names.
- The first column is the gene symbol (in
no particularalphabetical order).
is.NM.gene.txt: TRUE/FALSE flag for each gene indicating if the refSeq ID starts withNM_or not.- No header line.
- First column is the line number (in this file, add +1 for getting the line number in
genematrix_data.txt). - Second column is
TRUEorFALSE.
- Cohort data (in folder
cohortTables): A set of tab-separated files with data for each raw bioassay and the parent items it is derived from. Each file starts with a header line. Each row contains data for one raw bioassay. The first column (rba) is always the name of the raw bioassay. Columns ending with.A.are annotation columns. Date values are formatted asYYYY-MM-DDunless otherwise noted.cohortRawbioassay.txt: Data from the raw bioassay level. Columns:ID: Internal ID in BASEName: Name of raw bioassayPlatform: Name of platform (Sequencing)Raw.data.type: Name of raw data type (cufflinks)Has.data: Flag indicating if there is raw data for this raw bioassay or not (TRUE/FALSE)Db.spots: Number of raw data entriesArray.design: Name of the array designSoftware: Name of the software used to generate the raw dataImport.date: Date the raw data was createdAnalysisResult..A.DataFilesFolder..A.FPKM.tracking.file..F.: Path to theisoforms.fpkm_trackingfile in the BASE file system
cohortAligned.txt: Data from theAlignedSequencesparent item. Columns:ID: Internal ID in BASEName: Name of itemType: Type of item (AlignedSequences)Software: Name of the software used for alignmentRegistered: Date the item was registered in BASEAnalysisResult..A.DataFilesFolder..A.ALIGNED_PAIRS..A.READ_PAIRS_EXAMINED..A.READ_PAIR_DUPLICATES..A.FRACTION_DUPLICATION..A.FragmentSizeAvg..A.FragmentSizeStdev..A.
cohortMasked.txt: Data from theMaskedSequencesparent item. Columns:ID: Internal ID in BASEName: Name of itemType: Type of item (MaskedSequences)Software: Name of the software used for maskingRegistered: Date the item was registered in BASEPM_READS..A.
cohortMerged.txt: Data from theMergedSequencesparent item. Columns:ID: Internal ID in BASEName: Name of itemType: Type of item (MergedSequences)Physical.bioassays: Name of the physical bioassay (flow cell) used for sequencing. Comma-separated list if there is more than one.Software: Name of the software used for mergingRegistered: Date the item was registered in BASEAnalysisResult..A.DataFilesFolder..A.READS..A.PF_READS..A.ADAPTER_READS..A.PT_READS..A.FragmentSizeAvg..A.FragmentSizeStdev..A.
cohortSequencing.txt: Data from theSequencingRunparent item. Columns:- TODO
cohortLibrary.txt: Data from theLibraryparent item. Columns:ID: Internal ID in BASEName: Name of itemType: Type of item (Library)Protocol: Name of the library preparation protocolCreated: Date the library was createdTag: Name of the barcode used by the libraryBioplate: Name of the library plateBiowell.row: Row coordinate on the library plate (A-H)Biowell.column: Column coordinate on the library plate (1-12)QubitConc..A.
cohortRNA.txt: Data from theRNA/RNAQCparent item. Columns:ID: Internal ID in BASEName: Name of itemType: Type of item (RNA)Original.quantity..µg.: µg RNA that was extractedNDConc..A.ND260by230..A.ND260by280..A.QiacubeDate..A.QiacubeRunNo..A.QiacubePosition..A.RNAQC_last: RIN/RQS value from the latest quality control
cohortLysate.txt: Data from theLysateparent item. Columns:ID: Internal ID in BASEName: Name of itemType: Type of item (Lysate)Created: Date the lysate was createdOriginal.quantity..µg.: µg Lysate that was extractedParent.items: Name and used quantity from the specimenMultPieces..A.PartitionDate..A.
cohortSample.txt: Data from theSpecimenparent item. Columns:ID: Internal ID in BASEName: Name of itemType: Type of item (Specimen)Original.quantity..µg.: Weight of sample receivedCreated: Date the sample was created (operation date)ArrivalDate..A.BiopsyType..A.SpecimenType..A.Laterality..A.NofDeliveredTubes..A.NofPieces..A.OperatorDeliveryComment..A.OperatorPartitionComment..A.SamplingDateTime..A.RNALaterDateTime..A.LinkedSpecimen..A.
cohortCase.txt: Data from theCaseparent item (except INCA data). Columns:ID: Internal ID in BASEName: Name of itemType: Type of item (Case)Consent..A.ConsentDate..A.Laterality..A.
cohortPatient.txt: Data from thePatientparent item. Columns:ID: Internal ID in BASEName: Name of itemType: Type of item (Patient)Gender..A.Samples: Comma-separated list with all child item names (cases and blood)
cohortStained.txt: Data from theStainedparent item. To find the correct item we need to descend from the specimen level (Specimen -> Histology -> Stained) and select the one which hasGoodStain=TRUE. Columns:ID: Internal ID in BASEName: Name of itemType: Type of item (Case)CreatedRegisteredBioplateBiowell.rowBiowell.columnGoodStain..A.ScoreComplete..A.ScoreInvasiveCancer..A.ScoreInsituCancer..A.ScoreLymphocytes..A.ScoreStroma..A.ScoreFat..A.ScoreNormal..A.
cohortINCA.txt: Data from parent items (eg. Case) that have been imported from the INCA registry. Columns:IncaExportDate..A.- All other annotation types in the
INCAcategory. TheINCA_prefix is removed. No..A.is added in the header.
cohortSummaryTable.txt: A single table collecting some of the most useful information from the other tables. See the summary columns text file. The order of the columns in the file is not correct. They should have the same order as in the main data files.
- Subtype data (in folder
cohortTables/subtypeTables): Information generated by the R report scripts. We do not currently store this information in BASE, so it needs to be discussed how this should be done. The report plug-in could for example import the data from the R scripts as annotations.
- README files
- TODO
Change History (38)
comment:1 by , 9 years ago
| Description: | modified (diff) |
|---|
comment:2 by , 9 years ago
| Description: | modified (diff) |
|---|
comment:3 by , 9 years ago
| Description: | modified (diff) |
|---|
comment:4 by , 9 years ago
| Status: | new → assigned |
|---|
comment:5 by , 9 years ago
| Description: | modified (diff) |
|---|
comment:6 by , 9 years ago
| Description: | modified (diff) |
|---|
comment:7 by , 9 years ago
| Description: | modified (diff) |
|---|
comment:8 by , 9 years ago
| Description: | modified (diff) |
|---|
comment:9 by , 9 years ago
| Milestone: | Reggie v4.x → Reggie v4.5 |
|---|
comment:10 by , 9 years ago
(In [3932]) References #887: Release export wizard
Created the exporter plug-in (ReleaseExporterPlugin). This is a front-end for interacting with the user. The plug-in is installed in the "Item list" toolbar and asks for an output directory and if existing files should be overwritten or not.
The actual export is going to be implemented in the ReleaseExporter class. It is currently only able to collect the parameters and make some checks on the rawbioassays/array design.
comment:11 by , 9 years ago
comment:12 by , 9 years ago
(In [3934]) References #887: Release export wizard
Implemented exporter for th transcript data. Eg. all remaining files in the dataTables/transcriptDataTable directory.
Note that the current implementation only export data from the first raw bioassay. BASE 3.9 is needed to be able to export from all raw bioassays.
comment:13 by , 9 years ago
(In [3935]) References #887: Release export wizard
Started with the cohort exporters. The CohortItem is used for loading all information related to a single raw bioassays. It is used and then discarded in smaller batches to let garbage collection clean up memory.
The CohortWriter is an extension to the ReleaseWriter for writing cohort data. Each cohort data file need a subclass. The RawBioAssayWriter is the first such writer that write the raw bioassay data.
comment:14 by , 9 years ago
comment:15 by , 9 years ago
comment:16 by , 9 years ago
| Description: | modified (diff) |
|---|
comment:17 by , 9 years ago
comment:18 by , 9 years ago
comment:19 by , 9 years ago
| Description: | modified (diff) |
|---|
comment:20 by , 9 years ago
comment:21 by , 9 years ago
| Description: | modified (diff) |
|---|
comment:22 by , 9 years ago
| Description: | modified (diff) |
|---|
comment:23 by , 9 years ago
comment:24 by , 9 years ago
comment:25 by , 9 years ago
(In [3943]) References #887: Release export wizard
Started to implement the summary exporter. It has been implemented to copy columns from the other exporters. An extra complication is that the headers in the summary are different from the headers in the main data files. This is handled by adding the CohortHeader class which can hold two different headers. One for the main file and one for the summary.
So far, only the patient and case information is copied to the summary.
comment:26 by , 9 years ago
| Description: | modified (diff) |
|---|
comment:27 by , 9 years ago
comment:28 by , 9 years ago
comment:29 by , 9 years ago
| Description: | modified (diff) |
|---|
comment:30 by , 9 years ago
comment:31 by , 9 years ago
comment:32 by , 9 years ago
(In [3948]) References #887: Release export wizard
Implemented exporter for the genematrix_data.txt. There are (at least) two problems with the current approach:
- The database will probably choke on the "ORDER BY symbol" clause when used with a large set since this is not an indexed column. The query analyzer in Postgres shows that the join is done first and then the sort (for a data set with 1000 raw bioassays this is 100M rows to sort).
- The generated file is not compatible with the row numbering in
is.NM.gene.txtsince the raw bioassays doesn't contain data for all features.
{kind=link}
by , 9 years ago
| Attachment: | join-order-by-internal-id.png added |
|---|
{kind=link}
Join and sort by internal reporter id
by , 9 years ago
| Attachment: | no-join-order-by-internal-id.png added |
|---|
{kind=link}
No join sort by internal reporter id
comment:33 by , 9 years ago
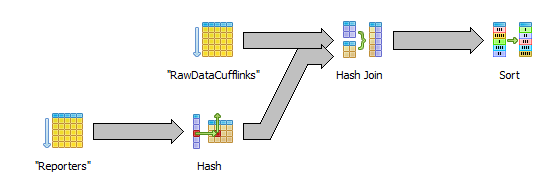
I have made some tests with different strategies for loading the raw data and producing the tidmatrix.* files. We want to avoid keeping too much data in memory at the same time which means that we must process data ordered by feature. Basically, we can choose to sort the data either by external id or internal id. The currently released files are sorted by external id.
I checked a few different queries in the PostgreSQL query planner.
Sort by external reporter id
select r.external_id, c.fpkm from "RawDataCufflinks" c inner join "Reporters" r on r.id=c.reporter_id order by r.external_id

Here raw data is retrieved via a sequential scan which is ok since we are going to need all in any case. The hash join to reporters is not a problem. The last sort step may turn out to be expensive though since it needs to sort 100M+ rows.
Sort by internal reporter id (keeping the join)
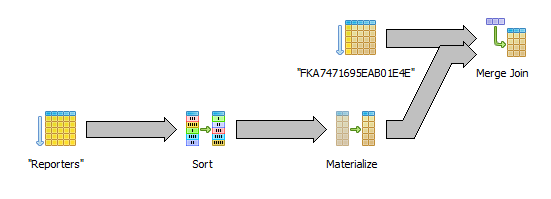
select r.external_id, c.fpkm from "RawDataCufflinks" c inner join "Reporters" r on r.id=c.reporter_id order by c.reporter_id

Here the raw data is retrieved in the desired order by using an index scan (FKA74...). The sort on reporter and the merge join should not too expensive.
Sort by internal id (without joining the reporters table)
select c.reporter_id, c.fpkm from "RawDataCufflinks" c order by c.reporter_id

This seems like the best alternative of them all. Only the sequential index scan is needed. We need to keep some reporter information in memory so we can get the external id from the internal id but that is not using very much memory.
comment:34 by , 9 years ago
| Description: | modified (diff) |
|---|
For efficient calculations it is desirable to process the data gene symbol by gene symbol. Thus, the data must come sorted in gene symbol order.